Whole-genome re-sequencing reveals genetic structure and selection signals of different populations in Megalobrama amblycephala
该研究发现团头鲂存在低遗传多样性及生长与耐低氧性状的选择信号,有助于其保护与育种工作。
Abstract
Characterization of genetic variants is crucial for understanding the molecular mechanisms behind selected traits and enhancing the rate of genetic gain through artificial selection. This study explored the genetic diversity and population structure of blunt snout bream Megalobrama amblycephala, a key freshwater fish in Chinese aquaculture. We conducted whole-genome re-sequencing of individuals from six populations, comprising three natural and three genetically improved strains, to assess genetic diversity, population structure and the impacts of selective breeding. We identified 17,009,590 high-quality biallelic single nucleotide polymorphism (SNP) loci. Genetic diversity assessments revealed low levels of polymorphism across populations, with genetically improved strains showing moderate to high genetic differentiation from their source populations. The study also highlighted runs of homozygosity (ROH) and analyzed linkage disequilibrium (LD) decay, indicating minimal recent inbreeding and diverse recombination events among populations. Selective sweep analyses uncovered significant selection loci associated with growth and hypoxia tolerance traits. Enrichment analyses identified genes involved in olfactory signal transduction, immune response, and metabolism pathways, which are closely related to these selective traits. These findings offer insights into the genetic basis of critical traits in M. amblycephala, contributing to the conservation of germplasm resources and supporting ongoing selective breeding efforts.
Introduction
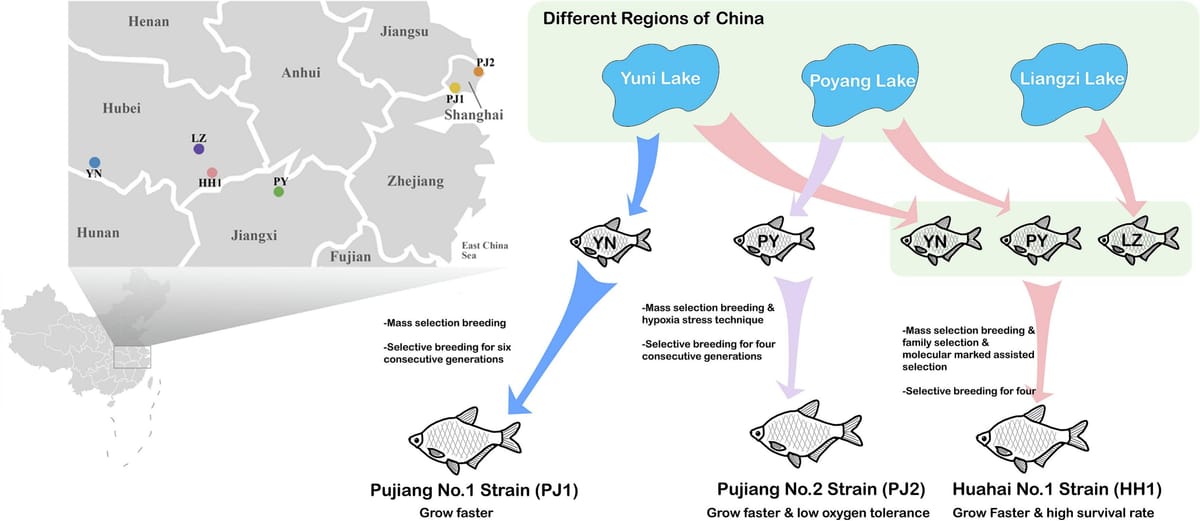
Aquaculture breeding provides numerous superior breeding varieties and creates greater production output, which is key to developing efficient aquaculture (Lhorente et al., 2019). Genetic diversity is a core concept in evolutionary biology and molecular ecology, playing a significant role in the complexity of organisms, the restoration of ecosystems, and the adaptation of species to diverse environmental conditions (Hughes et al., 2008; Shen and Yue, 2019). It is also a crucial theoretical foundation for genetic improvement. Studying genetic diversity not only reveals the evolutionary history and future directions of species or populations but also holds significant importance for the conservation of genetic resources, the development of biological resources, sustainable utilization, and maintaining global food security (Golden et al., 2021). According to Nguyen (Lind et al., 2012), conventional breeding methods applied to fish and shrimp may lead to an 8–12 % genetic gain with every generation, but currently, aquatic animals have only undergone limited genetic improvement or domestication (Brummett et al., 2004). Therefore, fully utilizing the wide genetic diversity in wild populations through breeding programs is very important for improving aquaculture productivity (Lind et al., 2012). Understanding the genetic structure and kinship of different groups is a crucial prerequisite for the rational and maximized use of genetic diversity resources. At the same time, with continuous progress in sequencing and bioinformatics, genomics can increasingly and conveniently be applied at various stages and in the domestication process of aquaculture species to optimize genetic improvement work (Houston et al., 2020).Whole-genome re-sequencing (WGRS) is a pivotal method for unveiling genetic variations and structural changes across different populations, aiding in the understanding of molecular breeding in animals and plants (Lin et al., 2022; Yu et al., 2018). It allows for the precise correlation of phenotypic variations with genetic diversity among populations, especially in the context of modern breeding (Wang et al., 2021a; Wang et al., 2021b). For instance, WGRS of 185 cigar tobacco plants elucidated population structure and identified genes associated with traits of artificial domestication, such as growth and disease resistance (Jiang et al., 2024). In the case of the Guizhou black goat, a significant meat production breed in Southwest China, Chang et al. (2024) conducted WGRS on core breeding populations to investigate genetic traits related to immunity, cold tolerance, and meat quality. Similarly, through the application of WGRS on Schizothorax o'connori, findings revealed minimal population differentiation alongside notable selection for energy metabolism and DNA repair in high-altitude populations (Gao et al., 2024). These studies collectively demonstrate the efficacy of WGRS in dissecting the genetic structure and identifying selection signatures across diverse populations, thereby enriching our understanding of population differentiation, kinship, and the genetic basis of important traits, which is crucial for conservation and breeding improvement efforts.Blunt snout bream (Megalobrama amblycephala), also known as Wuchang fish, is one of Chinese main freshwater species, which has always been favored by consumers for its delectable taste and ample nutritional value. The species was originally distributed in the middle Yangtze River and a few associated lakes, with Yuni Lake, Liangzi Lake, and Poyang Lake being the major sources (Li et al., 1991). To date, China has approved only three new varieties of M. amblycephala. These varieties have primarily been developed using conventional breeding methods and have not been extensively integrated into modern molecular genetic breeding techniques. Meanwhile, the research on the development of excellent varieties of M. amblycephala is insufficient, and concurrently, its germplasm resources are declining. Single nucleotide polymorphism (SNP) is the largest type of inherited genetic variation. Studies conducted in livestock have demonstrated that the emergence of a substantial number of SNP markers offers new opportunities for a more detailed assessment of genetic diversity (Eynard, 2018). In comparison to other marker systems, SNP markers possess distinct characteristics that make them well-suited for high-throughput and cost-effective genotyping platforms on a per data point basis (Saxena et al., 2012).In this study, a total of 180 M. amblycephala individuals were re-sequenced, comprising three natural populations and three genetically selected strains. SNPs were identified and used to investigate population genetic diversity and differentiation among three natural populations and three genetically selected strains. Furthermore, genomic selection signals in the genetically improved strains were analyzed in relation to the wild baseline population. Our aim is to facilitate further understanding of population structure and genetic diversity among different populations, which contributes to germplasm conservation and supports selective breeding program of M. amblycephala.
Access At ELSEVIER.
Explore scientific, technical, and medical research on ScienceDirect
Click to go